Ingesting Variants¶
This step describes how to ingest data into VarFish, that is
annotating variants and preparing them for import into VarFish
actually importing them into VarFish.
All of the steps below assume that you are running the Linux operating system. It might also work on Mac OS but is curently unsupported.
Variant Annotation¶
In order to import a VCF file with SNVs and small indels, the file has to be prepared for import into VarFish server. This is done using the Varfish Annotator software.
Installing the Annotator¶
The VarFish Annotator is written in Java and you can find the JAR on varfish-annotator Github releases page.
However, it is recommended to install it via bioconda.
For this, you first have to install bioconda as described in their manual.
Please ensure that you have the channels conda-forge, bioconda, and defaults set in the correct order as described in the bioconda installation manual.
A common pitfall is to forget the channel setup and subsequent failure to install varfish-annotator.
The next step is to install the varfish-annotator-cli package or create a conda environment with it.
# EITHER
$ conda install -y varfish-annotator-cli==0.14.0
# OR
$ conda create -y -n varfish-annotator varfish-annotator-cli==0.14.0
$ conda activate varfish-annotator
As a side remark, you might consider installing mamba first and then using mamba install and create in favour of conda install and create.
Obtaining the Annotator Data¶
The downloaded archive has a size of ~10 GB while the extracted data has a size of ~55 GB.
$ wget --no-check-certificate https://file-public.bihealth.org/transient/varfish/varfish-annotator-20201006.tar.gz{,.sha256}
$ sha256sum --check varfish-annotator-20201006.tar.gz.sha256
$ tar -xf varfish-annotator-20201006.tar.gz
$ ls varfish-annotator-20201006 | cat
hg19_ensembl.ser
hg19_refseq_curated.ser
hs37d5.fa
hs37d5.fa.fai
varfish-annotator-db-20201006.h2.db
Annotating VCF Files¶
First, obtain some tests data for annotation and later import into VarFish Server.
$ wget --no-check-certificate https://file-public.bihealth.org/transient/varfish/varfish-test-data-v0.22.2-20210212.tar.gz{,.sha256}
$ sha256sum --check varfish-test-data-v0.22.2-20210212.tar.gz.sha256
$ tar -xf varfish-test-data-v0.22.2-20210212.tar.gz.sha256
Next, you can use the varfish-annotator command:
1$ varfish-annotator \
2 -XX:MaxHeapSize=10g \
3 -XX:+UseConcMarkSweepGC \
4 annotate \
5 --db-path varfish-annotator-20201006/varfish-annotator-db-20191129.h2.db \
6 --ensembl-ser-path varfish-annotator-20201006/hg19_ensembl.ser \
7 --refseq-ser-path varfish-annotator-20201006/hg19_refseq_curated.ser \
8 --ref-path varfish-annotator-20201006/hs37d5.fa \
9 --input-vcf "INPUT.vcf.gz" \
10 --release "GRCh37" \
11 --output-db-info "FAM_name.db-info.tsv" \
12 --output-gts "FAM_name.gts.tsv" \
13 --case-id "FAM_name"
Let us disect this call.
The first three lines contain the code to the wrapper script and some arguments for the java binary to allow for enough memory when running.
1$ varfish-annotator \
2 -XX:MaxHeapSize=10g \
3 -XX:+UseConcMarkSweepGC \
The next lines use the annotate sub command and provide the needed paths to the database files needed for annotation.
The .h2.db file contains information from variant databases such as gnomAD and ClinVar.
The .ser file are transcript databases used by the Jannovar library.
The .fa file is the path to the genome reference file used.
While only release GRCh37/hg19 is supported, using a file with UCSC-style chromosome names having chr prefixes would also work.
4annotate \ --db-path varfish-annotator-20201006/varfish-annotator-db-20191129.h2.db \ --ensembl-ser-path varfish-annotator-20201006/hg19_ensembl.ser \ --refseq-ser-path varfish-annotator-20201006/hg19_refseq_curated.ser \ --ref-path varfish-annotator-20201006/hs37d5.fa \
The following lines provide the path to the input VCF file, specify the release name (must be GRCh37) and the name of the case as written out.
This could be the name of the index patient, for example.
9--input-vcf "INPUT.vcf.gz" \ --release "GRCh37" \ --case-id "index" \
The last lines
12--output-db-info "FAM_name.db-info.tsv" \ --output-gts "FAM_name.gts.tsv"
After the program terminates, you should create gzip files for the created TSV files and md5 sum files for them.
$ gzip -c FAM_name.db-info.tsv >FAM_name.db-info.tsv.gz
$ md5sum FAM_name.db-info.tsv.gz >FAM_name.db-info.tsv.gz.md5
$ gzip -c FAM_name.gts.tsv >FAM_name.gts.tsv.gz
$ md5sum FAM_name.gts.tsv.gz >FAM_name.gts.tsv.gz.md5
The next step is to import these files into VarFish server. For this, a PLINK PED file has to be provided. This is a tab-separated values (TSV) file with the following columns:
family name
individul name
father name or
0for foundermother name or
0for foundersex of individual,
1for male,2for female,0if unknowndisease state of individual,
1for unaffected,2for affected,0if unknown
For example, a trio would look as follows:
FAM_index index father mother 2 2
FAM_index father 0 0 1 1
FAM_index mother 0 0 2 1
while a singleton could look as follows:
FAM_index index 0 0 2 1
Note that you have to link family individuals with pseudo entries that have no corresponding entry in the VCF file. For example, if you have genotypes for two siblings but none for the parents:
FAM_index sister father mother 2 2
FAM_index broth father mother 2 2
FAM_index father 0 0 1 1
FAM_index mother 0 0 2 1
Variant Import¶
As a prerequisite you need to install the VarFish command line interface (CLI) Python app varfish-cli.
You can install it from PyPi with pip install varfish-cli or from Bioconda with conda install varfish-cli.
Second, you need to create a new API token as described in API Token Management.
Then, setup your Varfish CLI configuration file ~/.varfishrc.toml as:
[global]
varfish_server_url = "https://varfish.example.com/"
varfish_api_token = "XXX"
Now you can import the data that you imported above.
You will also find some example files in the test-data directory.
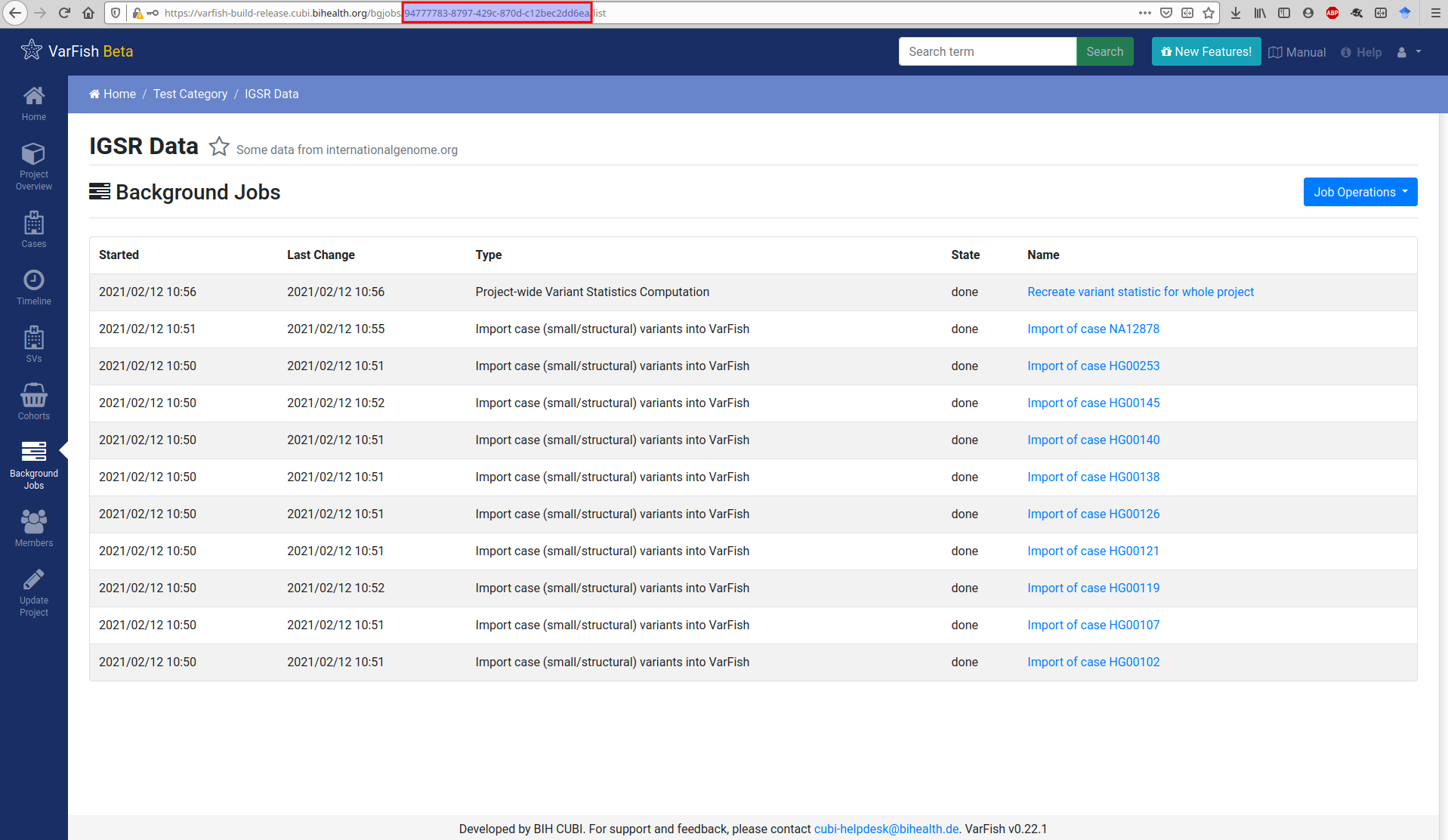
For the import you will also need the project UUID. You can get this from the URLs in VarFish that list project properties. The figure below shows this for the background job list but this also works for the project details view.
$ varfish-cli --no-verify-ssl case create-import-info --resubmit \
94777783-8797-429c-870d-c12bec2dd6ea \
test-data/tsv/HG00102-N1-DNA1-WES1/*.{tsv.gz,.ped}
When executing the import as shown above, you have to specify:
a pedigree file with suffix
.ped,a genotype annotation file as generated by
varfish-annotatorending in.gts.tsv.gz,a database info file as generated by
varfish-annotatorending in.db-infos.tsv.gz.
Optionally, you can also specify a TSV file with BAM quality control metris ending in .bam-qc.tsv.gz.
Currently, the format is not properly documented yet but documentation and supporting tools are forthcoming.
Running the import command through VarFish CLI will create a background import job as shown below. Once the job is done, the created or updated case will appear in the case list.
Undocumented¶
The following needs to be properly documented here:
Preparation of the BAM QC file that has the information about duplication rate etc. You can have a look at the
*.bam-qc.tsv.gzfiles below thetest-datadirectory.